| Weight | 1 lbs |
|---|---|
| Dimensions | 9 × 5 × 2 in |
| host | rabbit |
| isotype | IgG |
| clonality | polyclonal |
| concentration | 1 mg/mL |
| applications | IHC, WB |
| reactivity | human |
| available sizes | 10 µL, 100 µL |


rabbit anti-SOD1 polyclonal antibody 1026
Price range: $100.00 through $2,600.00
Antibody summary
- Rabbit polyclonal to Superoxide Dismutase 1 (SOD)
- Suitable for: WB,IHC
- Reacts with: Hu presumed Guinea Pig, Ms, Dog, Rat
- Isotype: IgG
- 100 µL, 10 µL
rabbit anti- sod1 polyclonal antibody 1026
| antibody |
|---|
| Database link: human P00441 mouse P08228 rat P07632 |
| Tested applications WB,IHC |
| Recommended dilutions WB: 1:2000-10000, IHC: 1:500-2000, Epitope retrieval with citrate buffer pH 6.0 is recommended for FFPE tissue sections. |
| Immunogen Between 104 and C-terminus |
| Size and concentration 100µL and 1 mg/mL |
| Form liquid |
| Storage Instructions 2-8°C for short term, for longer term at -20°C. Avoid freeze / thaw cycles. |
| Storage buffer Tris-citrate/phosphate buffer, pH 7 to 8 containing 0.09% Sodium Azide |
| Purity affinity purified |
| Clonality polyclonal |
| Isotype IgG |
| Compatible secondaries goat anti-rabbit IgG, H&L chain specific, peroxidase conjugated, conjugated polyclonal antibody 9512 goat anti-rabbit IgG, H&L chain specific, biotin conjugated polyclonal antibody 2079 goat anti-rabbit IgG, H&L chain specific, FITC conjugated polyclonal antibody 7863 goat anti-rabbit IgG, H&L chain specific, Cross Absorbed polyclonal antibody 2371 goat anti-rabbit IgG, H&L chain specific, biotin conjugated polyclonal antibody, crossabsorbed 1715 goat anti-rabbit IgG, H&L chain specific, FITC conjugated polyclonal antibody, crossabsorbed 1720 |
| Isotype control Rabbit polyclonal - Isotype Control |
| target relevance |
|---|
| Homo sapiens SOD1 Superoxide dismutase [Cu-Zn] |
| Protein names Superoxide dismutase [Cu-Zn] |
| Alternative names Hydrogen sulfide oxidase, Superoxide dismutase 1 |
| Gene names SOD1 |
| Protein family Belongs to the Cu-Zn superoxide dismutase family |
| Function Destroys radicals which are normally produced within the cells and which are toxic to biological systems (PubMed:24140062). Catalyzes the oxidation of hydrogen sulfide (H2S) to sulfate, playing an important role in detoxifying H2S and limiting the accumulation of reactive sulfur species (RSS) such as persulfides and polysulfides (PubMed:36630448) |
| Catalytic activity 2 superoxide + 2 H(+) = H2O2 + O2 hydrogen sulfide + 2 O2 = sulfate + H(+) |
| Subcellular location Cytoplasm, Nucleus |
| Structure Homodimer; non-disulfide-linked (By similarity). Homodimerization may take place via the ditryptophan cross-link at Trp-33. Heterodimer with SOD1 (PubMed:31292775). The heterodimer CCS:SOD1 interacts with SLC31A1; this heterotrimer is Cu(1+)-mediated and its maintenance is regulated through SOD1 activation (PubMed:31292775). Interacts with DAOA; the interaction is direct (PubMed:30037290) |
| Post-translational modification Unlike wild-type protein, the pathogenic variants ALS1 Arg-38, Arg-47, Arg-86 and Ala-94 are polyubiquitinated by RNF19A leading to their proteasomal degradation. The pathogenic variants ALS1 Arg-86 and Ala-94 are ubiquitinated by MARCH5 leading to their proteasomal degradation The ditryptophan cross-link at Trp-33 is responsible for the non-disulfide-linked homodimerization. Such modification might only occur in extreme conditions and additional experimental evidence is required Palmitoylation helps nuclear targeting and decreases catalytic activity Succinylation, adjacent to copper catalytic site, probably inhibits activity. Desuccinylation by SIRT5 enhances activity |
| Involvement in disease Amyotrophic lateral sclerosis 1 A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. Spastic tetraplegia and axial hypotonia, progressive An autosomal recessive, neurologic disorder characterized by loss of motor abilities in the first year of life, after which severe, progressive spastic tetraparesis develops. Affected individuals have severe axial hypotonia, hyperekplexia, hypertonia, and myokymia, reflecting upper motor neuron involvement. Cognitive development may be affected. |
| Keywords 3D-structure, Acetylation, Amyotrophic lateral sclerosis, Antioxidant, Copper, Cytoplasm, Direct protein sequencing, Disease variant, Disulfide bond, Lipoprotein, Metal-binding, Neurodegeneration, Nucleus, Oxidoreductase, Palmitate, Phosphoprotein, Proteomics identification, Reference proteome, Ubl conjugation, Zinc |
| Sequence MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ |
| UniProt accession: P00441 |
Data
 |
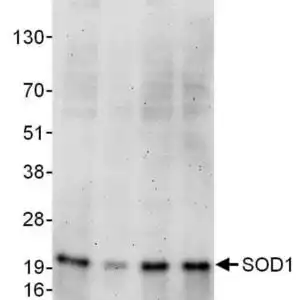
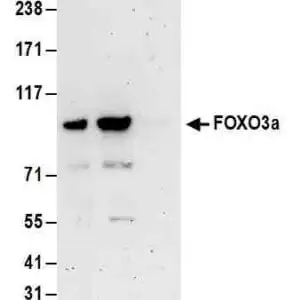
| Detection of human SOD1 by western blot. Samples: Whole cell lysate from HeLa (15 and 50 µg), HEK293T (T; 50 µg) and Jurkat (J; 50 µg) cells. Antibodies: Affinity purified rabbit anti-SOD1 antibody #1026 used for WB at 0.1 µg/ml. Detection: Chemiluminescence with an exposure time of 3 minutes. |
 |
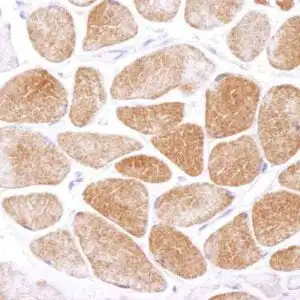
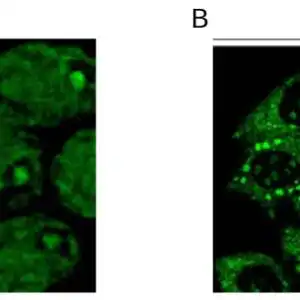
| Detection of human SOD1 by immunohistochemistry. Sample: FFPE section of human osteosarcoma. Antibody: Affinity purified rabbit anti-SOD1 (#1026) used at a dilution of 1:1,000 (1 µg/ml). Detection: DAB |
FAQ & Publications
Frequently Asked Questions
What species does the rabbit anti-SOD1 polyclonal antibody 1026 react with?
This antibody reacts with human, presumed guinea pig, mouse, dog, and rat species.
Which applications has the rabbit anti-SOD1 polyclonal antibody 1026 been validated for?
It has been tested and validated for use in Western blotting (WB) and immunohistochemistry (IHC) applications.
What are the recommended antibody dilutions for Western blot and immunohistochemistry?
For Western blot, the recommended dilution range is 1:2000 to 1:10000. For immunohistochemistry, a dilution range of 1:500 to 1:2000 is advised, with epitope retrieval using citrate buffer at pH 6.0 for formalin-fixed paraffin-embedded tissue sections.
How should the rabbit anti-SOD1 polyclonal antibody 1026 be stored to maintain stability?
For short-term storage, keep the antibody at 2-8°C. For long-term storage, it should be stored at -20°C, avoiding repeated freeze-thaw cycles.
What is the concentration and form of the rabbit anti-SOD1 polyclonal antibody 1026 provided?
The antibody is supplied as a liquid at a concentration of 1 mg/mL, available in 10 µL and 100 µL sizes.
Publications
| pmid | title | authors | citation |
|---|---|---|---|
| We haven't added any publications to our database yet. | |||
Published literature highly relevant to the biological target of this product and referencing this antibody or clone are retrieved from the PubMed database provided by the United States National Library of Medicine at the National Institutes of Health.
Protocols
| relevant to this product |
|---|
| Western blot IHC |
Documents
| Batch Number | QC File | SDS |
|---|---|---|
| To view batch-specific Safety Datasheets and Quality Certificates associated with your account, please Log In. | ||
Only logged in customers who have purchased this product may leave a review.

Reviews
There are no reviews yet.