| Weight | 1 lbs |
|---|---|
| Dimensions | 9 × 5 × 2 in |
| host | mouse |
| isotype | IgG2b |
| clonality | monoclonal |
| concentration | concentrate, predilute |
| applications | IHC |
| reactivity | human |
| available size | 0.1 mL, 0.5 mL, 1 mL concentrated, 7 mL prediluted |

mouse anti-p63 monoclonal antibody (ZM70) 6315
Price range: $160.00 through $528.00
Antibody summary
- Mouse monoclonal to p63
- Suitable for: Immunohistochemistry (formalin-fixed, paraffin-embedded tissues)
- Reacts with: Human
- Isotype:IgG2b
- Control: Skin or prostate
- Visualization: Nuclear
- 0.1, 0.5, 1.0 mL concentrated, 7 mL prediluted
mouse anti-p63 monoclonal antibody ZM70 6315
| target relevance |
|---|
| Homo sapiens TP63 Tumor protein 63 |
| Protein names Tumor protein 63 |
| Alternative names Chronic ulcerative stomatitis protein, Keratinocyte transcription factor KET, Transformation-related protein 63, Tumor protein p73-like, p40, p51 |
| Gene names TP63 |
| Protein family Belongs to the p53 family |
| Function Acts as a sequence specific DNA binding transcriptional activator or repressor. The isoforms contain a varying set of transactivation and auto-regulating transactivation inhibiting domains thus showing an isoform specific activity. Isoform 2 activates RIPK4 transcription. May be required in conjunction with TP73/p73 for initiation of p53/TP53 dependent apoptosis in response to genotoxic insults and the presence of activated oncogenes. Involved in Notch signaling by probably inducing JAG1 and JAG2. Plays a role in the regulation of epithelial morphogenesis. The ratio of DeltaN-type and TA*-type isoforms may govern the maintenance of epithelial stem cell compartments and regulate the initiation of epithelial stratification from the undifferentiated embryonal ectoderm. Required for limb formation from the apical ectodermal ridge. Activates transcription of the p21 promoter |
| Subcellular location Nucleus |
| Structure Binds DNA as a homotetramer. Isoform composition of the tetramer may determine transactivation activity. Isoforms Alpha and Gamma interact with HIPK2. Interacts with SSRP1, leading to stimulate coactivator activity. Isoform 1 and isoform 2 interact with WWP1. Interacts with PDS5A. Isoform 5 (via activation domain) interacts with NOC2L |
| Post-translational modification May be sumoylated Ubiquitinated. Polyubiquitination involves WWP1 and leads to proteasomal degradation of this protein |
| Involvement in disease Acro-dermato-ungual-lacrimal-tooth syndrome A form of ectodermal dysplasia. Ectodermal dysplasia defines a heterogeneous group of disorders due to abnormal development of two or more ectodermal structures. ADULT syndrome involves ectrodactyly, syndactyly, finger- and toenail dysplasia, hypoplastic breasts and nipples, intensive freckling, lacrimal duct atresia, frontal alopecia, primary hypodontia and loss of permanent teeth. ADULT syndrome differs significantly from EEC3 syndrome by the absence of facial clefting. Inheritance is autosomal dominant. Ankyloblepharon-ectodermal defects-cleft lip/palate An autosomal dominant condition characterized by congenital ectodermal dysplasia with coarse, wiry, sparse hair, dystrophic nails, slight hypohidrosis, scalp infections, ankyloblepharon filiform adnatum, maxillary hypoplasia, hypodontia and cleft lip/palate. Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 A form of ectodermal dysplasia, a heterogeneous group of disorders due to abnormal development of two or more ectodermal structures. It is an autosomal dominant syndrome characterized by ectrodactyly of hands and feet, ectodermal dysplasia and facial clefting. Split-hand/foot malformation 4 A limb malformation involving the central rays of the autopod and presenting with syndactyly, median clefts of the hands and feet, and aplasia and/or hypoplasia of the phalanges, metacarpals, and metatarsals. Some patients have been found to have intellectual disability, ectodermal and craniofacial findings, and orofacial clefting. Limb-mammary syndrome Characterized by ectrodactyly, cleft palate and mammary-gland abnormalities. Rapp-Hodgkin syndrome A form of ectodermal dysplasia, a heterogeneous group of disorders due to abnormal development of two or more ectodermal structures. Characterized by the combination of anhidrotic ectodermal dysplasia, cleft lip, and cleft palate. The clinical syndrome is comprised of a characteristic facies (narrow nose and small mouth), wiry, slow-growing, and uncombable hair, sparse eyelashes and eyebrows, obstructed lacrimal puncta/epiphora, bilateral stenosis of external auditory canals, microsomia, hypodontia, cone-shaped incisors, enamel hypoplasia, dystrophic nails, and cleft lip/cleft palate. RHS inheritance is autosomal dominant. Orofacial cleft 8 A birth defect consisting of cleft lips with or without cleft palate. Cleft lips are associated with cleft palate in two-third of cases. A cleft lip can occur on one or both sides and range in severity from a simple notch in the upper lip to a complete opening in the lip extending into the floor of the nostril and involving the upper gum. Premature ovarian failure 21 A form of premature ovarian failure, an ovarian disorder defined as the cessation of ovarian function under the age of 40 years. It is characterized by oligomenorrhea or amenorrhea, in the presence of elevated levels of serum gonadotropins and low estradiol. POF21 inheritance is autosomal dominant. |
| Keywords 3D-structure, Activator, Alternative promoter usage, Alternative splicing, Apoptosis, Developmental protein, Disease variant, DNA-binding, Ectodermal dysplasia, Isopeptide bond, Metal-binding, Notch signaling pathway, Nucleus, Premature ovarian failure, Proteomics identification, Reference proteome, Transcription, Transcription regulation, Ubl conjugation, Zinc |
| Sequence MNFETSRCATLQYCPDPYIQRFVETPAHFSWKESYYRSTMSQSTQTNEFLSPEVFQHIWD FLEQPICSVQPIDLNFVDEPSEDGATNKIEISMDCIRMQDSDLSDPMWPQYTNLGLLNSM DQQIQNGSSSTSPYNTDHAQNSVTAPSPYAQPSSTFDALSPSPAIPSNTDYPGPHSFDVS FQQSSTAKSATWTYSTELKKLYCQIAKTCPIQIKVMTPPPQGAVIRAMPVYKKAEHVTEV VKRCPNHELSREFNEGQIAPPSHLIRVEGNSHAQYVEDPITGRQSVLVPYEPPQVGTEFT TVLYNFMCNSSCVGGMNRRPILIIVTLETRDGQVLGRRCFEARICACPGRDRKADEDSIR KQQVSDSTKNGDGTKRPFRQNTHGIQMTSIKKRRSPDDELLYLPVRGRETYEMLLKIKES LELMQYLPQHTIETYRQQQQQQHQHLLQKQTSIQSPSSYGNSSPPLNKMNSMNKLPSVSQ LINPQQRNALTPTTIPDGMGANIPMMGTHMPMAGDMNGLSPTQALPPPLSMPSTSHCTPP PPYPTDCSIVSFLARLGCSSCLDYFTTQGLTTIYQIEHYSMDDLASLKIPEQFRHAIWKG ILDHRQLHEFSSPSHLLRTPSSASTVSVGSSETRGERVIDAVRFTLRQTISFPPRDEWND FNFDMDARRNKQQRIKEEGE |
| UniProt accession: Q9H3D4 |
Data
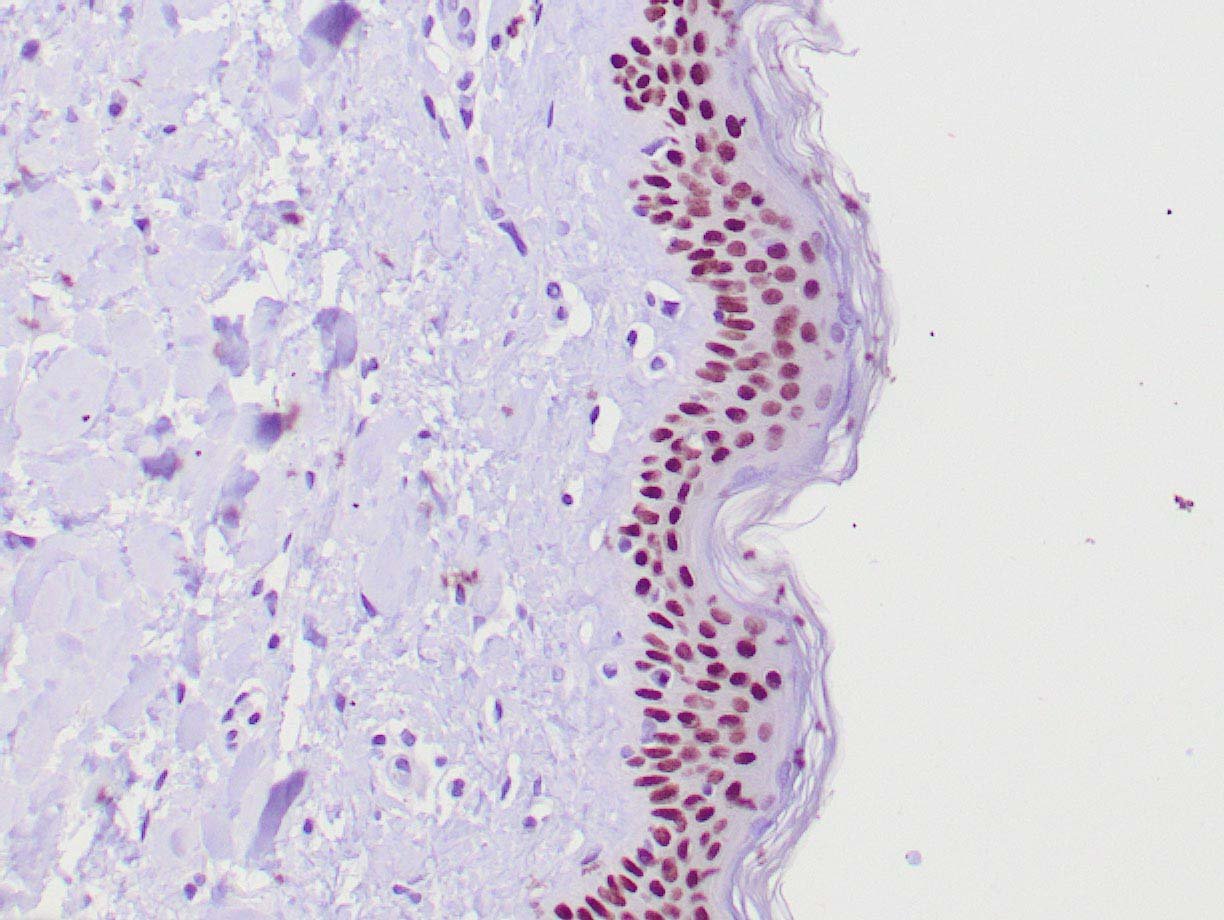 |
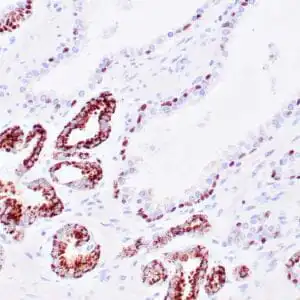
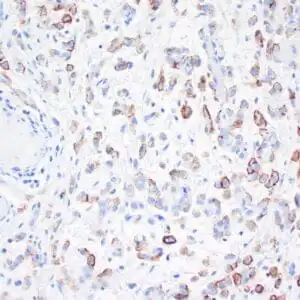
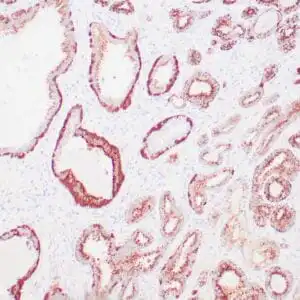
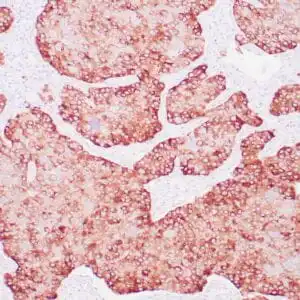
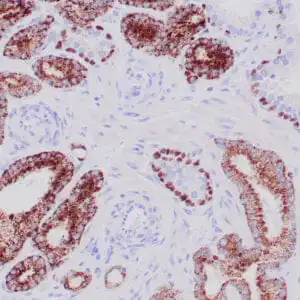
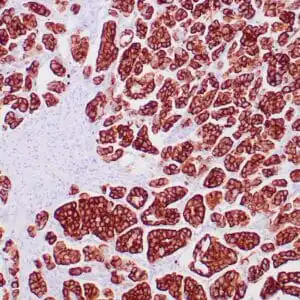
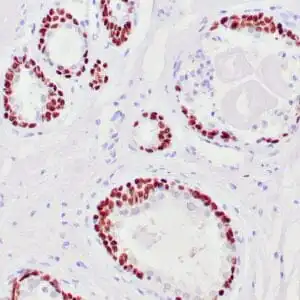
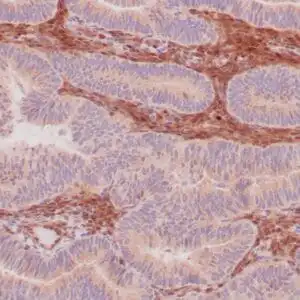
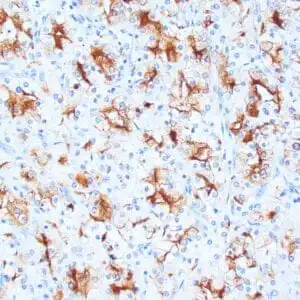
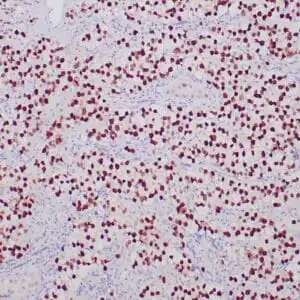
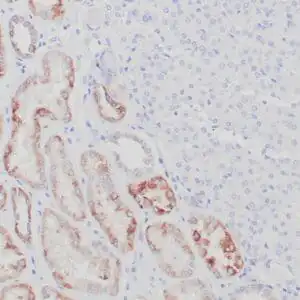
| Human skin stained with anti-p63 using peroxidase conjugate and DAB chromogen. Note nuclear staining of basal cells. |
FAQ & Publications
Frequently Asked Questions
What species does the mouse anti-p63 monoclonal antibody (ZM70) 6315 react with?
This antibody is reactive with human tissue samples.
What are the recommended storage conditions for the mouse anti-p63 monoclonal antibody (ZM70) 6315?
For short-term storage, keep the antibody at 2-8°C. For long-term storage, store it at -20°C and avoid repeated freeze/thaw cycles.
Which applications is the mouse anti-p63 monoclonal antibody (ZM70) 6315 validated for?
This antibody is suitable for immunohistochemistry (IHC) on formalin-fixed, paraffin-embedded human tissues.
Publications
| pmid | title | authors | citation |
|---|---|---|---|
| We haven't added any publications to our database yet. | |||
Published literature highly relevant to the biological target of this product and referencing this antibody or clone are retrieved from the PubMed database provided by the United States National Library of Medicine at the National Institutes of Health.
Protocols
| relevant to this product |
|---|
| IHC |
Documents
| Batch Number | QC File | SDS |
|---|---|---|
| To view batch-specific Safety Datasheets and Quality Certificates associated with your account, please Log In. | ||
Only logged in customers who have purchased this product may leave a review.
Reviews
There are no reviews yet.