| Weight | 1 lbs |
|---|---|
| Dimensions | 9 × 5 × 2 in |
| host | mouse |
| isotype | IgG2b/κ |
| clonality | monoclonal |
| concentration | concentrate, predilute |
| applications | IHC |
| reactivity | human |
| available size | 0.1 mL, 0.5 mL, 1 mL concentrated, 7 mL prediluted |

mouse anti-Cytokeratin 14 monoclonal antibody (ZM372) 6136
Price range: $160.00 through $528.00
Antibody summary
- Mouse monoclonal to Cytokeratin 14
- Suitable for: Immunohistochemistry (formalin-fixed, paraffin-embedded tissues)
- Reacts with: Human
- Isotype:IgG2b/κ
- Control: Skin, tonsil or squamous cell carcinoma
- Visualization: Cytoplasmic
- 0.1, 0.5, 1.0 mL concentrated, 7 mL prediluted
mouse anti-Cytokeratin 14 monoclonal antibody ZM372 6136
| target relevance |
|---|
| Homo sapiens KRT14 Keratin, type I cytoskeletal 14 |
| Protein names Keratin, type I cytoskeletal 14 |
| Alternative names Cytokeratin-14, Keratin-14 |
| Gene names KRT14 |
| Protein family Belongs to the intermediate filament family |
| Function The nonhelical tail domain is involved in promoting KRT5-KRT14 filaments to self-organize into large bundles and enhances the mechanical properties involved in resilience of keratin intermediate filaments in vitro |
| Subcellular location Cytoplasm, Nucleus |
| Structure Heterotetramer of two type I and two type II keratins (By similarity). Forms a disulfide-linked heterodimer (via 2B domains) with KRT5 (via 2B domains) (PubMed:22705788, PubMed:24940650). Forms a heterodimer with KRT1; the interaction is more abundant in the absence of KRT5 (By similarity). Interacts with PLEC isoform 1C, when in a heterodimer with KRT5 (PubMed:24940650). Interacts with TRADD and with keratin filaments (PubMed:11684708). Associates with other type I keratins (PubMed:11724817). Interacts with EPPK1 (By similarity). Interacts with KLHL24 (PubMed:27798626). Interacts with PKP1 (via N-terminus) and PKP2 (PubMed:10852826) |
| Post-translational modification A disulfide bond is formed between rather than within filaments and promotes the formation of a keratin filament cage around the nucleus Ubiquitinated by the BCR(KLHL24) E3 ubiquitin ligase complex |
| Involvement in disease Epidermolysis bullosa simplex 1A, generalized severe A form of epidermolysis bullosa simplex, a group of skin fragility disorders characterized by skin blistering due to cleavage within the basal layer of keratinocytes, and erosions caused by minor mechanical trauma. There is a broad spectrum of clinical severity ranging from minor blistering on the feet, to subtypes with extracutaneous involvement and a lethal outcome. EBS1A is an autosomal dominant form characterized by generalized intraepidermal skin blistering that begins and is very prominent at birth. EBS1A may be life-threatening in the first year of life. Tendency to blistering diminishes in adolescence. Epidermolysis bullosa simplex 1C, localized A form of epidermolysis bullosa simplex, a group of skin fragility disorders characterized by skin blistering due to cleavage within the basal layer of keratinocytes, and erosions caused by minor mechanical trauma. There is a broad spectrum of clinical severity ranging from minor blistering on the feet, to subtypes with extracutaneous involvement and a lethal outcome. EBS1C is an autosomal dominant form with intraepidermal blistering mainly restricted to hands and feet beginning in infancy. Nails may be thick and dystrophic. Epidermolysis bullosa simplex 1B, generalized intermediate A form of epidermolysis bullosa simplex, a group of skin fragility disorders characterized by skin blistering due to cleavage within the basal layer of keratinocytes, and erosions caused by minor mechanical trauma. There is a broad spectrum of clinical severity ranging from minor blistering on the feet, to subtypes with extracutaneous involvement and a lethal outcome. EBS1B is an autosomal dominant form characterized by generalized intraepidermal blistering beginning at birth. The tendency to blistering diminishes in adolescence, when it may become localized to hands and feet. Epidermolysis bullosa simplex 1D, generalized, intermediate or severe, autosomal recessive A form of epidermolysis bullosa simplex, a group of skin fragility disorders characterized by skin blistering due to cleavage within the basal layer of keratinocytes, and erosions caused by minor mechanical trauma. There is a broad spectrum of clinical severity ranging from minor blistering on the feet, to subtypes with extracutaneous involvement and a lethal outcome. EBS1D is an autosomal recessive form characterized by blistering beginning at birth or early childhood. In some patients hands and feet are primarily affected, and in others blistering anywhere on the body may occur. In some patients the condition improves with age. Naegeli-Franceschetti-Jadassohn syndrome A rare autosomal dominant form of ectodermal dysplasia. The cardinal features are absence of dermatoglyphics (fingerprints), reticular cutaneous hyperpigmentation (starting at about the age of 2 years without a preceding inflammatory stage), palmoplantar keratoderma, hypohidrosis with diminished sweat gland function and discomfort provoked by heat, nail dystrophy, and tooth enamel defects. Dermatopathia pigmentosa reticularis A rare ectodermal dysplasia characterized by lifelong persistent reticulate hyperpigmentation, non-cicatricial alopecia, and nail dystrophy. Variable features include adermatoglyphia, hypohidrosis or hyperhidrosis, and palmoplantar hyperkeratosis. |
| Keywords 3D-structure, Coiled coil, Cytoplasm, Disease variant, Disulfide bond, Ectodermal dysplasia, Epidermolysis bullosa, Intermediate filament, Keratin, Nucleus, Palmoplantar keratoderma, Phosphoprotein, Proteomics identification, Reference proteome, Ubl conjugation |
| Sequence MTTCSRQFTSSSSMKGSCGIGGGIGGGSSRISSVLAGGSCRAPSTYGGGLSVSSSRFSSG GACGLGGGYGGGFSSSSSSFGSGFGGGYGGGLGAGLGGGFGGGFAGGDGLLVGSEKVTMQ NLNDRLASYLDKVRALEEANADLEVKIRDWYQRQRPAEIKDYSPYFKTIEDLRNKILTAT VDNANVLLQIDNARLAADDFRTKYETELNLRMSVEADINGLRRVLDELTLARADLEMQIE SLKEELAYLKKNHEEEMNALRGQVGGDVNVEMDAAPGVDLSRILNEMRDQYEKMAEKNRK DAEEWFFTKTEELNREVATNSELVQSGKSEISELRRTMQNLEIELQSQLSMKASLENSLE ETKGRYCMQLAQIQEMIGSVEEQLAQLRCEMEQQNQEYKILLDVKTRLEQEIATYRRLLE GEDAHLSSSQFSSGSQSSRDVTSSSRQIRTKVMDVHDGKVVSTHEQVLRTKN |
| UniProt accession: P02533 |
Data
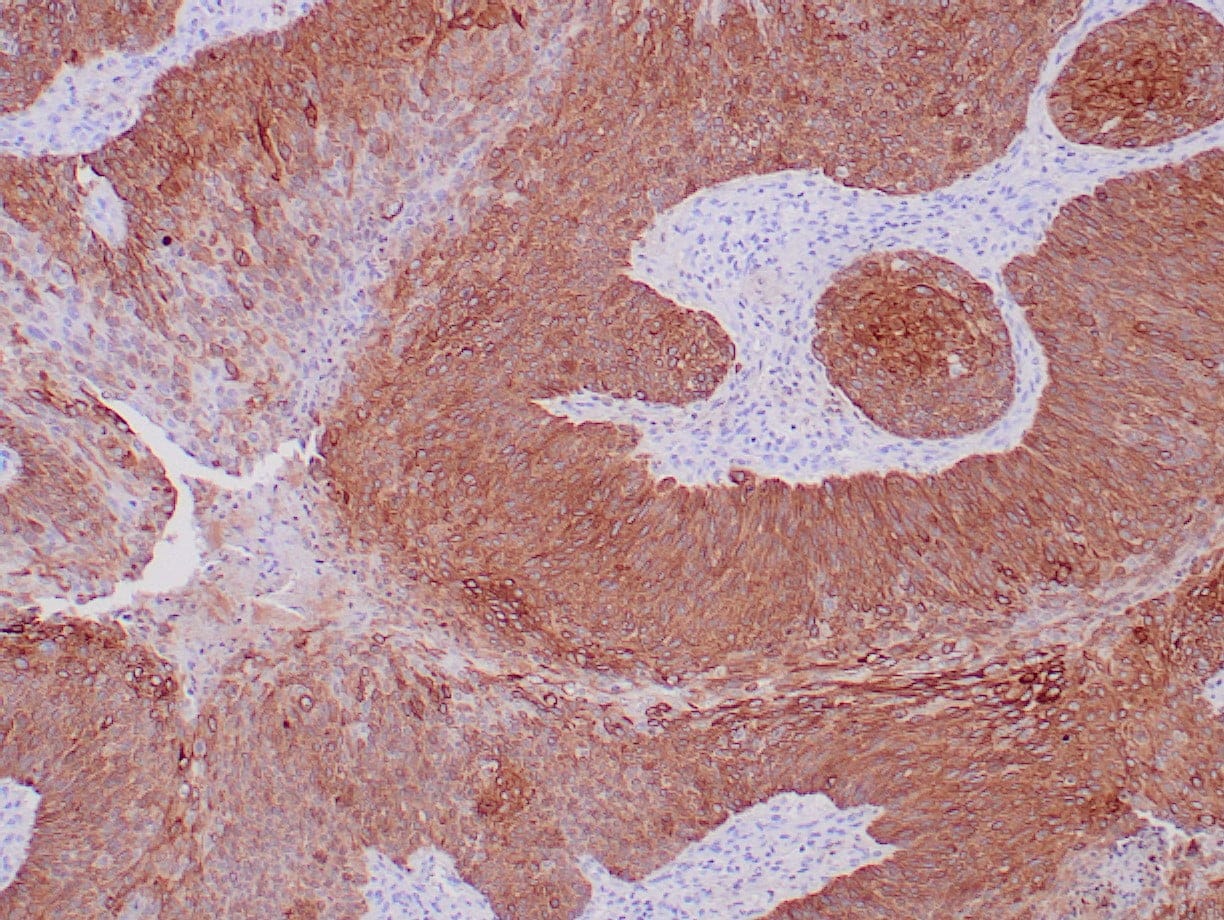 |
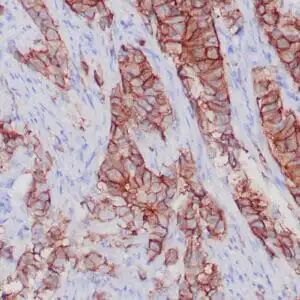
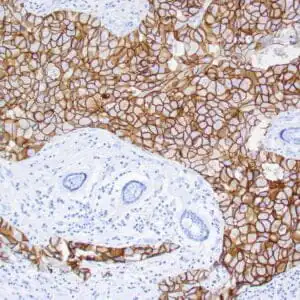
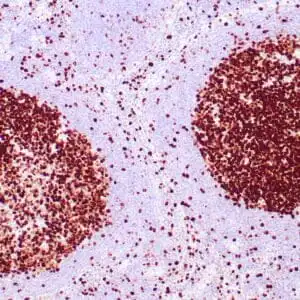
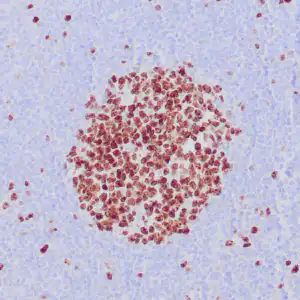
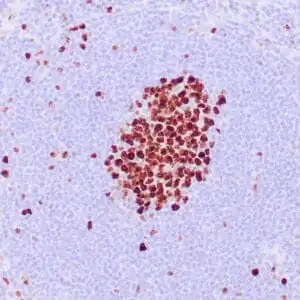
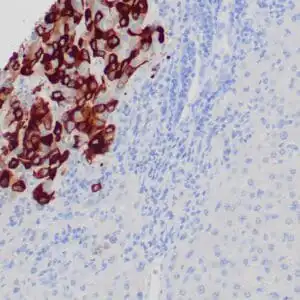
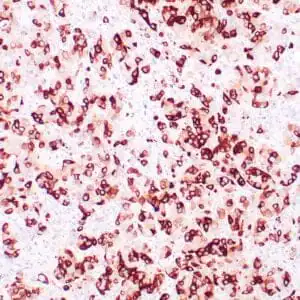
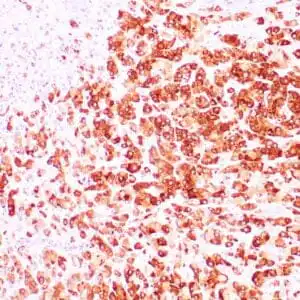
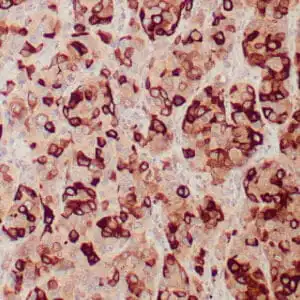
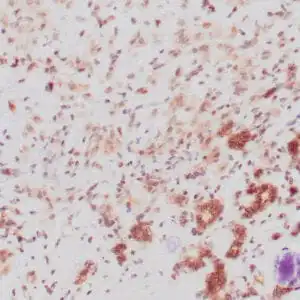
| Formalin-fixed, paraffin-embedded human squamous cell carcinoma stained with CK14 using peroxidase-conjugate and DAB chromogen. Note the cytoplasmic staining of squamous cells |
FAQ & Publications
Frequently Asked Questions
What species does the mouse anti-Cytokeratin 14 monoclonal antibody (ZM372) 6136 specifically react with?
This antibody specifically reacts with human Cytokeratin 14.
Which applications is this monoclonal antibody validated for?
The antibody is suitable and tested for Immunohistochemistry (IHC) on formalin-fixed, paraffin-embedded tissues.
What are the recommended dilutions for using the concentrated form of this antibody in IHC?
The recommended dilution for the concentrated antibody is between 1:100 and 1:200 for immunohistochemistry applications.
How should the mouse anti-Cytokeratin 14 monoclonal antibody be stored to maintain stability?
For short-term storage, keep the antibody at 2-8°C. For longer-term storage, it should be kept at -20°C, avoiding repeated freeze/thaw cycles.
What controls are suggested when using this anti-Cytokeratin 14 antibody in immunohistochemistry?
Positive control tissues recommended include skin, tonsil, or squamous cell carcinoma samples.
Publications
| pmid | title | authors | citation |
|---|---|---|---|
| We haven't added any publications to our database yet. | |||
Published literature highly relevant to the biological target of this product and referencing this antibody or clone are retrieved from the PubMed database provided by the United States National Library of Medicine at the National Institutes of Health.
Protocols
| relevant to this product |
|---|
| IHC |
Documents
| Batch Number | QC File | SDS |
|---|---|---|
| To view batch-specific Safety Datasheets and Quality Certificates associated with your account, please Log In. | ||
Only logged in customers who have purchased this product may leave a review.
Reviews
There are no reviews yet.